Hội chứng DiGeorge có cách điều trị?
 Những cái kết thương tâm do bệnh dại đến từ sự chủ quan
Những cái kết thương tâm do bệnh dại đến từ sự chủ quan Bất ngờ với công dụng của trái thù lù, trước mọc dại giờ là vị thuốc quý được
Bất ngờ với công dụng của trái thù lù, trước mọc dại giờ là vị thuốc quý đượcHội chứng DiGeorge được coi là một trong những hội chứng di truyền phổ biến, chỉ đứng sau hội chứng Down.
Trong dân số nói chung, ước tính tỷ lệ ảnh hưởng là 1 trên 4.000 ca sinh. Vậy hội chứng DiGeorge có điều trị được không?
1. Hội chứng DiGeorge là gì?
Hội chứng DiGeorge, còn được gọi là hội chứng mất đoạn 22q11.2 là một rối loạn phức tạp. Hội chứng DiGeorge có thể là một hội chứng nhưng thực chất nó là một tập hợp các đặc điểm lâm sàng và một loạt các tình trạng bệnh lý ảnh hưởng đến nhiều hệ thống khác nhau trong cơ thể.
Mức độ nghiêm trọng và biểu hiện của các triệu chứng có thể khác nhau đáng kể giữa những cá nhân bị ảnh hưởng, ngay cả trong cùng một gia đình, do đó, việc chẩn đoán cũng như điều trị trở thành một thách thức lớn.
Phát hiện, chẩn đoán sớm, kết hợp với phương pháp chăm sóc y tế phù hợp, đánh giá tim mạch, đánh giá miễn dịch, hỗ trợ phát triển là rất quan trọng để cải thiện chất lượng cuộc sống của bệnh nhân mắc hội chứng DiGeorge.
Bệnh nhân mắc hội chứng DiGeorge có thể có một số khiếm khuyết, kém phát triển ở một số cấu trúc, chức năng cơ thể do mất một đoạn nhiễm sắc thể 22.
Thiểu sản tuyến ức hoặc bất sản tuyến ức là một trong những khiếm khuyết quan trọng nhất liên quan đến hội chứng này, dẫn đến rối loạn chức năng hệ thống miễn dịch khiến tỷ lệ nhiễm trùng cao. Tế bào lympho T, là cốt lõi của phản ứng hệ thống miễn dịch của con người chống lại các tác nhân gây bệnh, phụ thuộc rất nhiều vào tuyến ức để phát triển, trưởng thành.

Hội chứng DiGeorge, còn được gọi là hội chứng mất đoạn 22q11.2 là một rối loạn phức tạp.
Ngoài khiếm khuyết tuyến ức, bệnh nhân mắc hội chứng DiGeorge cũng có thể bị thiểu sản hoặc rối loạn chức năng tuyến cận giáp đóng vai trò điều hòa nồng độ canxi trong máu. Nồng độ canxi thấp hoặc hạ canxi máu có thể gây co giật, chuột rút cơ và các biến chứng thần kinh khác nếu không được điều trị.
Các dấu hiệu khác của hội chứng DiGeorge là các khuyết tật tim bẩm sinh, bất thường ở vòm miệng và đặc điểm trên khuôn mặt, sự chậm phát triển và các rối loạn tâm thần, cùng nhiều dấu hiệu khác.
2. Cách chẩn đoán hội chứng DiGeorge
Chẩn đoán thường được thực hiện dựa trên các dấu hiệu, triệu chứng xuất hiện khi sinh hoặc phát triển ngay sau khi sinh. Chẩn đoán thường có thể được xác nhận bằng các xét nghiệm di truyền gọi là lai ghép huỳnh quang tại chỗ (FISH).
Nguyên nhân phổ biến nhất của hội chứng này là do một đoạn nhiễm sắc thể 22 bị mất. Một tỷ lệ nhỏ bệnh nhân không có khiếm khuyết di truyền được chẩn đoán dựa trên các đặc điểm lâm sàng và bằng cách loại trừ chẩn đoán các hội chứng khác.
Do đặc điểm rất khác nhau nên hội chứng này thường dễ bị bỏ sót hoặc chẩn đoán sai. Việc điều trị thường phụ thuộc vào mức độ nghiêm trọng của các triệu chứng. Đối với những gia đình muốn có con, giải pháp tốt nhất là tư vấn di truyền.

Phát hiện, điều trị sớm các khuyết tật và các biến chứng khác, nhiều bệnh nhân mắc hội chứng DiGeorge có thể có chất lượng cuộc sống tốt cho đến khi trưởng thành.
3. Các biện pháp điều trị hội chứng DiGeorge
Hiện tại chưa có phương pháp điều trị cụ thể cho hội chứng này. Phương pháp điều trị tương ứng chủ yếu dựa vào triệu chứng của người bệnh và phải được các bác sĩ thuộc nhiều chuyên khoa cùng đánh giá, chăm sóc, bao gồm:
- Sau khi sinh, cần đánh giá hô hấp và oxy hóa máu, tiến hành kiểm tra siêu âm tim, nếu cần thiết nên tiến hành điều trị bằng phẫu thuật sớm.
- Nếu hệ thống miễn dịch bị suy giảm, cần đánh giá sự cần thiết của việc ghép tủy càng sớm càng tốt, đồng thời cân nhắc thời điểm tiêm vaccine, sử dụng kháng sinh dự phòng tùy theo tình hình để giảm tỷ lệ nhiễm trùng nghiêm trọng.
- Bệnh nhân sứt môi, hở hàm ếch nên đánh giá thời điểm can thiệp phẫu thuật càng sớm càng tốt để tránh ảnh hưởng đến tình trạng ăn uống.
- Trẻ sơ sinh, thanh thiếu niên nên thường xuyên theo dõi nồng độ canxi trong máu. Nếu phải phẫu thuật hoặc xảy ra nhiễm trùng nặng, nồng độ canxi trong máu cũng cần được theo dõi. Nếu xảy ra tình trạng hạ canxi máu, cần bổ sung canxi và vitamin D để phòng ngừa co giật. Cũng cần thường xuyên theo dõi nồng độ canxi trong máu và nước tiểu sau khi dùng thuốc.
- Can thiệp vào việc điều trị phục hồi chức năng càng sớm càng tốt, như rèn luyện sức mạnh cơ bắp, trị liệu ngôn ngữ và đánh giá xem có các tình trạng như mất tập trung và tự kỷ khi thích hợp hay không.
- Việc chăm sóc tâm lý ở tuổi trưởng thành cũng cần được quan tâm thường xuyên. Cần đánh giá thường xuyên sự tăng trưởng và phát triển của bệnh nhân trong thời thơ ấu. Nếu có tầm vóc thấp bé, cần chẩn đoán sớm tình trạng khó ăn hoặc thiếu hụt hormone tăng trưởng và đưa ra phương pháp điều trị tương ứng. Tuy nhiên, bạn cần chú ý kiểm soát cân nặng ở tuổi trưởng thành để tránh hội chứng chuyển hóa liên quan đến béo phì.
- Sắp xếp khám siêu âm thận và sàng lọc thính giác để phát hiện sớm các bất thường về phát triển liên quan và chú ý đến các triệu chứng liên quan đến bệnh tự miễn.
Nhìn chung, tuổi thọ của bệnh nhân mắc hội chứng DiGeorge phụ thuộc vào mức độ nghiêm trọng của các triệu chứng, sự hiện diện của các rối loạn y tế liên quan và khả năng tiếp cận dịch vụ chăm sóc y tế cũng như sự hỗ trợ phù hợp.
Nhờ sự ra đời của các liệu pháp và can thiệp y tế, phát hiện, điều trị sớm các khuyết tật tim bẩm sinh, rối loạn chức năng hệ thống miễn dịch và các biến chứng khác, nhiều bệnh nhân mắc hội chứng DiGeorge có thể có chất lượng cuộc sống tốt cho đến khi trưởng thành.
Sức khỏe cô gái có nửa trái tim vượt ngoài dự đoán của bác sĩ
Khi chào đời, Amy Roberts chỉ có một nửa trái tim còn hoạt động nên được tiên lượng không thể sống lâu.
Trước đó, các bác sĩ đã khuyên mẹ của Amy nên bỏ thai ở tuần thứ 20 sau khi phát hiện tình trạng dị tật phức tạp. Nhưng bố mẹ cô từ chối, muốn cho đứa con chưa chào đời của mình một cơ hội chiến đấu.
Amy hiện sống với cha mẹ, có một anh trai và một chị gái. Cô chào đời với cả hai động mạch tim đều bị lệch hướng và có một tâm thất trái đôi. Đây là khiếm khuyết rất hiếm gặp, khi bên phải của tim nhỏ và kém phát triển, nguồn cung cấp máu chuyển sang bên trái.

Amy đã tập gym được 4 năm nay, giúp tăng cường sức khỏe tim mạch. Ảnh: Express
Theo Express, Amy đã trải qua ba cuộc phẫu thuật tim hở khi còn nhỏ và sự mạo hiểm của gia đình cô đã được đền đáp. Lần mổ đầu tiên diễn ra khi Amy mới 5 ngày tuổi. Các bác sĩ đặt ống dẫn lưu vào tĩnh mạch chính để giữ lại mạng sống cho cô.
Sau đó, Amy tiếp tục những cuộc phẫu thuật phức tạp hơn khi 2 và 7 tuổi. Mặc dù ghét môn thể dục ở trường nhưng Amy vẫn áp dụng chế độ tập luyện vừa phải giúp ích cho trái tim.
Ở tuổi 22, Amy đã trở thành người sống lâu nhất ở Anh chỉ với nửa trái tim. Kể từ khi cô tham gia tập gym cách đây 4 năm, chức năng tim đã được cải thiện rất nhiều. Các chuyên gia nhận định sau này, Amy có thể sẽ không cần cấy ghép tim.
Amy, đến từ Worsley, Greater Manchester (Anh), cho biết: "Trước đây, các bác sĩ nói rằng nếu tôi sống sót, tôi sẽ cần cấy ghép tim khi 20 tuổi. Nhưng trong lần khám cuối cùng, họ nói với tôi rằng chức năng tim của tôi đã tốt hơn và khỏe hơn nhiều. Có thể tôi sẽ không cần ghép tim cho đến khi sang tuổi 40, đó là một tin tuyệt vời".
Cô nói thêm: "Tôi bắt đầu tập luyện khi 18 tuổi, vì lúc đó tôi rất ốm yếu. Lúc đầu, tôi không thể nâng được một chiếc tạ nào, nhưng tôi bắt đầu từ từ và dần dần nâng cao bản thân. Tôi tập luyện 5 lần/tuần và tôi cũng là huấn luyện viên cá nhân. Tôi rất vui vì đã rèn luyện được sức mạnh của mình như thế này, giúp tôi có một sức sống mới. Bây giờ, tôi cảm thấy rất tuyệt".
Người phát ngôn của Tiny Tickers, tổ chức từ thiện dành cho trẻ sơ sinh mắc bệnh tim, cho biết: "Amy đang tiến triển rất tốt dù từng bị tiên lượng không có khả năng sống sót cao. Hầu hết bệnh nhân mắc bệnh này đều không thể qua được tuổi thiếu niên".
Tin liên quan
 Hội chứng thiên thần: những đứa trẻ xinh đẹp dù đau đớn vẫn luôn tươi cười
Hội chứng thiên thần: những đứa trẻ xinh đẹp dù đau đớn vẫn luôn tươi cười Em bé sinh ra với tay chân khác biệt, chỉ mong được đi dép
Em bé sinh ra với tay chân khác biệt, chỉ mong được đi dép Dấu hiệu sớm bệnh tim bẩm sinh ở trẻ
Dấu hiệu sớm bệnh tim bẩm sinh ở trẻ Bé gái bị thoát vị hoành kèm theo dị tật phổi biệt lập
Bé gái bị thoát vị hoành kèm theo dị tật phổi biệt lập Trẻ em bị viêm nướu, chảy máu chân răng do đâu?
Trẻ em bị viêm nướu, chảy máu chân răng do đâu? Tìm ra nguyên nhân suy giảm chức năng hệ miễn dịch
Tìm ra nguyên nhân suy giảm chức năng hệ miễn dịch 3 loại trà thảo dược giúp sáng mắt
3 loại trà thảo dược giúp sáng mắt
 Cách xoa bóp khi đau mỏi cổ gáy
Cách xoa bóp khi đau mỏi cổ gáy Điều trị bàn chân bẹt như thế nào?
Điều trị bàn chân bẹt như thế nào? Dấu hiệu cảnh báo bệnh lao phổi
Dấu hiệu cảnh báo bệnh lao phổi Loại quả Việt 'nữ hoàng vitamin C' đa công dụng ngỡ ngàng vì cực dễ tìm
Loại quả Việt 'nữ hoàng vitamin C' đa công dụng ngỡ ngàng vì cực dễ tìmTiêu điểm
 Thời điểm ăn đồ ngọt tốt nhất để kiểm soát đường huyết
Thời điểm ăn đồ ngọt tốt nhất để kiểm soát đường huyết 9 đồ uống giàu magie giúp ngủ ngon, giảm mệt mỏi
9 đồ uống giàu magie giúp ngủ ngon, giảm mệt mỏi Bé trai sốc nhiễm khuẩn, suy thận cấp sau khi uống sữa hộp pha sẵn
Bé trai sốc nhiễm khuẩn, suy thận cấp sau khi uống sữa hộp pha sẵn Bé gái 15 tháng tuổi bị ô tô cán qua hồi phục kỳ diệu
Bé gái 15 tháng tuổi bị ô tô cán qua hồi phục kỳ diệu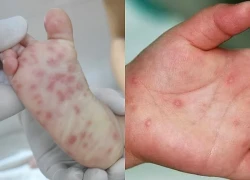 Ca tay chân miệng tăng vọt, chuyên gia khuyến cáo 5 nguyên tắc phòng bệnh
Ca tay chân miệng tăng vọt, chuyên gia khuyến cáo 5 nguyên tắc phòng bệnh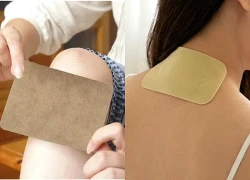 Cứ đau là dán cao, xoa dầu nóng: 3 rủi ro nhiều người bỏ qua
Cứ đau là dán cao, xoa dầu nóng: 3 rủi ro nhiều người bỏ qua 4 lầm tưởng về đường huyết và cách kiểm soát hiệu quả theo chuyên gia
4 lầm tưởng về đường huyết và cách kiểm soát hiệu quả theo chuyên gia Triệu chứng mắc biến thể Covid-19 'Ve sầu'
Triệu chứng mắc biến thể Covid-19 'Ve sầu'Tin đang nóng
 Lee Min Ho kết hôn vào tháng 5
Lee Min Ho kết hôn vào tháng 5 Bích Phương không thể ngăn Tăng Duy Tân nữa rồi!
Bích Phương không thể ngăn Tăng Duy Tân nữa rồi! Vì "không đủ vâng lời", Song Hye Kyo bị nhà chồng cũ hắt hủi ra mặt
Vì "không đủ vâng lời", Song Hye Kyo bị nhà chồng cũ hắt hủi ra mặt Mỹ dỡ bỏ trừng phạt Tổng thống lâm thời Venezuela
Mỹ dỡ bỏ trừng phạt Tổng thống lâm thời Venezuela Không tin được đây là ca sĩ tỷ phú giàu nhất thế giới, nhan sắc xuống cấp đến mức này rồi sao?
Không tin được đây là ca sĩ tỷ phú giàu nhất thế giới, nhan sắc xuống cấp đến mức này rồi sao? Dư luận Hàn Quốc phẫn nộ sau vụ đạo diễn bị đánh dẫn đến chết não
Dư luận Hàn Quốc phẫn nộ sau vụ đạo diễn bị đánh dẫn đến chết não Bất ngờ với giá trị số vàng Lệ Quyên nhận được trong ngày sinh nhật
Bất ngờ với giá trị số vàng Lệ Quyên nhận được trong ngày sinh nhật Tổng thống Trump lần đầu phát biểu trước toàn quốc về xung đột Iran
Tổng thống Trump lần đầu phát biểu trước toàn quốc về xung đột IranTin mới nhất
Cấp cứu người hoại tử chi nguy kịch bằng kỹ thuật "lần đầu tiên ở Việt Nam"

Phát hiện quan trọng về vai trò của sắt trong quá trình sản xuất insulin
Thấy con sau 27 năm mờ nhòe, người mẹ bật khóc khi "được sinh ra lần nữa"
Bộ Y tế thu hồi lô thuốc Aceclofenac STELLA 100mg
Cảnh báo chủng virus EV71 độc lực cao khiến bệnh tay chân miệng trở nặng nhanh
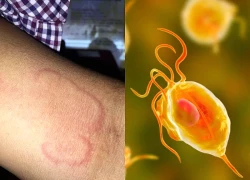
Bệnh ký sinh trùng nhiều người dương tính khi xét nghiệm ở Khánh Hòa nguy hiểm mức nào nếu không điều trị sớm?
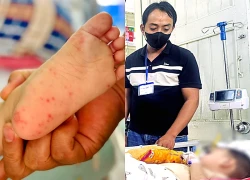
Giám sát các 'điểm nóng' tay chân miệng tại phường Bình Hưng Hòa

Nghiên cứu 460.000 người: Tác dụng bất ngờ từ 2 - 3 tách cà phê mỗi ngày

Bộ phận 'bẩn nhất' của gà nhưng là món khoái khẩu của nhiều người

5 loại thực phẩm người trên 40 tuổi nên tránh để bảo vệ sức khỏe

5 chế độ ăn lành mạnh có thể giúp kéo dài tuổi thọ

Không phải cá hồi: 3 loại cá 'bé xíu' giá rẻ đầy chợ Việt nhưng 'bổ ngang hàng'
Có thể bạn quan tâm

Trấn Thành: Xem MV Quỳnh Anh Shyn tưởng của CL hay Lady Gaga
Nhạc việt
00:28:02 03/04/2026
Trung Quốc mới có 1 phim ngôn tình gây nghiện cực mạnh: Nam chính trời sinh làm tổng tài, đẹp đôi với cả thế giới
Phim châu á
00:24:45 03/04/2026
Việt Nam mới có 1 mỹ nhân cổ trang xinh đáo để: Hào quang từ đầu đến chân, lả lướt mướt mắt vô cùng
Phim việt
00:07:50 03/04/2026
Viện trưởng Khánh Thi không còn sợ hãi, con trai Cục trưởng Xuân Bắc gây sốt
Sao việt
23:59:32 02/04/2026
Đặng Tụy Văn từng sống như 'người hầu' để nuôi ước mơ làm diễn viên
Sao châu á
23:52:57 02/04/2026
Sharon Stone thừa nhận trả giá đắt vì cảnh 'nóng' trong 'Bản năng gốc'
Sao âu mỹ
23:50:03 02/04/2026
Phương Anh Đào nói về phim mới của Đoàn Thiên Ân
Hậu trường phim
23:18:34 02/04/2026Chevron bác bỏ cáo buộc thao túng giá xăng
Thế giới
21:36:55 02/04/2026
Tử vi 12 cung hoàng đạo 2/4: Kim Ngưu một bước lên mây, dễ bị ghen tị
Trắc nghiệm
21:27:35 02/04/2026
Không chỉ ASIAD hay LCK, Ruler hoàn toàn có thể đối diện cái kết tệ hơn nữa
Mọt game
21:03:01 02/04/2026
 Cô gái 10X du học về làm nghề tảo mộ thuê, nhận lương 15 triệu
Cô gái 10X du học về làm nghề tảo mộ thuê, nhận lương 15 triệu Cảnh báo tình mẹ con có nguy cơ sứt mẻ sau khi loạt tranh này được công bố!
Cảnh báo tình mẹ con có nguy cơ sứt mẻ sau khi loạt tranh này được công bố! Hội mỹ nữ Việt được báo Trung khen lên tận mây xanh từ 6 - 7 năm trước, giờ ra sao?
Hội mỹ nữ Việt được báo Trung khen lên tận mây xanh từ 6 - 7 năm trước, giờ ra sao? Ô tô dừng ngược chiều, người đàn ông ở Hà Nội hành hung tài xế xe máy
Ô tô dừng ngược chiều, người đàn ông ở Hà Nội hành hung tài xế xe máy Người đàn ông rơi từ cầu Vĩnh Tuy tử vong: Một chi tiết đầy xót xa và ám ảnh tại hiện trường
Người đàn ông rơi từ cầu Vĩnh Tuy tử vong: Một chi tiết đầy xót xa và ám ảnh tại hiện trường Khán giả xót xa cho Lê Phương
Khán giả xót xa cho Lê Phương Cẩm Ly tuyên bố đặc biệt khi Hoài Lâm đi hát trở lại
Cẩm Ly tuyên bố đặc biệt khi Hoài Lâm đi hát trở lại Đến ra mắt nhà bạn trai, cô gái chết lặng khi nhìn thấy "bố chồng tương lai", vội vã rời đi
Đến ra mắt nhà bạn trai, cô gái chết lặng khi nhìn thấy "bố chồng tương lai", vội vã rời đi Lan truyền bức ảnh với nội dung "Lọ Lem về ra mắt nhà thiếu gia Sầm Nhất ở Tây Ninh": Phản ứng của chính chủ
Lan truyền bức ảnh với nội dung "Lọ Lem về ra mắt nhà thiếu gia Sầm Nhất ở Tây Ninh": Phản ứng của chính chủ Phạm Băng Băng đi bán mặt nạ ở siêu thị: Nhan sắc thật khiến đám đông tròn mắt, đứng hình kinh ngạc
Phạm Băng Băng đi bán mặt nạ ở siêu thị: Nhan sắc thật khiến đám đông tròn mắt, đứng hình kinh ngạc